AMCG Sequence Variant Details#
This section describes additional details on our implementation of ACMG classification for sequence variants.
Data#
The following datasources are used in the classification of sequence variants.
Data |
Tool / Datasource |
Original Datasource |
Transcripts |
RefSeq / ENSEMBL |
|
Variant Effect Predictions |
N/A |
|
Variant Frequencies |
annonars |
gnomAD exomes, genomes, mtDNA |
chrMT scores, annotations |
annonars |
MITOMAP, MitoTip, MitImpact, MtSNPscore |
gene annotations |
annonars |
ClinGen, GCD, gene2phenotype, GenCC, PanelApp, DOMINO |
protein annotations: domains, repeats, mutations |
UCSC genome browser |
UniProt |
References#
Literature with direct ACMG / ACGS / ClinGen relationship
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015 May;17(5):405-24. doi: 10.1038/gim.2015.30. Epub 2015 Mar 5. PMID: 25741868; PMCID: PMC4544753.
ClinGen Sequence Variant Interpretation Work Group. Recommendations for ACMG/AMP guideline criteria code modifications nomenclature. November 10, 2017.
Whiffin N, Minikel E, Walsh R, O’Donnell-Luria AH, Karczewski K, Ing AY, Barton PJR, Funke B, Cook SA, MacArthur D, Ware JS. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet Med. 2017 Oct;19(10):1151-1158. doi: 10.1038/gim.2017.26. Epub 2017 May 18. PMID: 28518168; PMCID: PMC5563454.
Abou Tayoun AN, Pesaran T, DiStefano MT, Oza A, Rehm HL, Biesecker LG, Harrison SM; ClinGen Sequence Variant Interpretation Working Group (ClinGen SVI). Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat. 2018 Nov;39(11):1517-1524. doi: 10.1002/humu.23626. Epub 2018 Sep 7. PMID: 30192042; PMCID: PMC6185798.
Biesecker LG, Harrison SM; ClinGen Sequence Variant Interpretation Working Group. The ACMG/AMP reputable source criteria for the interpretation of sequence variants. Genet Med. 2018 Dec;20(12):1687-1688. doi: 10.1038/gim.2018.42. PMID: 29543229; PMCID: PMC6709533.
Ghosh R, Harrison SM, Rehm HL, Plon SE, Biesecker LG; ClinGen Sequence Variant Interpretation Working Group. Updated recommendation for the benign stand-alone ACMG/AMP criterion. Hum Mutat. 2018 Nov;39(11):1525-1530. doi: 10.1002/humu.23642. PMID: 30311383; PMCID: PMC6188666.
ClinGen Sequence Variant Interpretation Work Group. SVI Recommendation for in trans Criterion (PM3) - Version 1.0 2019.
Brnich SE, Abou Tayoun AN, Couch FJ, Cutting GR, Greenblatt MS, Heinen CD, Kanavy DM, Luo X, McNulty SM, Starita LM, Tavtigian SV, Wright MW, Harrison SM, Biesecker LG, Berg JS; Clinical Genome Resource Sequence Variant Interpretation Working Group. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med. 2019 Dec 31;12(1):3. doi: 10.1186/s13073-019-0690-2. PMID: 31892348; PMCID: PMC6938631.
Ellard S, Baple EL, Berry I, Forrester N, Turnbull C, Owens M, Eccles D, Abbs S, Scott R, Deans Z. ACGS best practice guidelines for variant classification 2019. 2019.
ClinGen Sequence Variant Interpretation Work Group. SVI Recommendation for Absence/Rarity (PM2) - Version 1.0 2020.
Ellard S, Baple EL, Callaway A, Berry I, Forrester N, Turnbull C, Owens M, Eccles DM, Abbs S, Scott R, Deans ZC, Lester T, Campbell J, Newman WG, Ramsden S, McMullan DJ. ACGS Best Practice Guidelines for Variant Classification in Rare Disease 2020. 2020.
McCormick EM, Lott MT, Dulik MC, Shen L, Attimonelli M, Vitale O, Karaa A, Bai R, Pineda-Alvarez DE, Singh LN, Stanley CM, Wong S, Bhardwaj A, Merkurjev D, Mao R, Sondheimer N, Zhang S, Procaccio V, Wallace DC, Gai X, Falk MJ. Specifications of the ACMG/AMP standards and guidelines for mitochondrial DNA variant interpretation. Hum Mutat. 2020 Dec;41(12):2028-2057. doi: 10.1002/humu.24107. Epub 2020 Nov 10. PMID: 32906214; PMCID: PMC7717623.
Tavtigian SV, Harrison SM, Boucher KM, Biesecker LG. Fitting a naturally scaled point system to the ACMG/AMP variant classification guidelines. Hum Mutat. 2020 Oct;41(10):1734-1737. doi: 10.1002/humu.24088. Epub 2020 Aug 30. PMID: 32720330; PMCID: PMC8011844.
ClinGen Sequence Variant Interpretation Work Group. SVI Recommendation for De Novo Criteria (PS2 & PM6) - Version 1.1 2021.
DiStefano MT, Goehringer S, Babb L, Alkuraya FS, Amberger J, Amin M, Austin-Tse C, Balzotti M, Berg JS, Birney E, Bocchini C. The gene curation coalition: a global effort to harmonize gene-disease evidence resources. Genetics in Medicine. 2022 Aug 1;24(8):1732-42.
Pejaver V, Byrne AB, Feng BJ, Pagel KA, Mooney SD, Karchin R, O’Donnell-Luria A, Harrison SM, Tavtigian SV, Greenblatt MS, Biesecker LG, Radivojac P, Brenner SE; ClinGen Sequence Variant Interpretation Working Group. Calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for PP3/BP4 criteria. Am J Hum Genet. 2022 Dec 1;109(12):2163-2177. doi: 10.1016/j.ajhg.2022.10.013. Epub 2022 Nov 21. PMID: 36413997; PMCID: PMC9748256.
Walker LC, Hoya M, Wiggins GAR, Lindy A, Vincent LM, Parsons MT, Canson DM, Bis-Brewer D, Cass A, Tchourbanov A, Zimmermann H, Byrne AB, Pesaran T, Karam R, Harrison SM, Spurdle AB; ClinGen Sequence Variant Interpretation Working Group. Using the ACMG/AMP framework to capture evidence related to predicted and observed impact on splicing: Recommendations from the ClinGen SVI Splicing Subgroup. Am J Hum Genet. 2023 Jul 6;110(7):1046-1067. doi: 10.1016/j.ajhg.2023.06.002. Epub 2023 Jun 22. PMID: 37352859; PMCID: PMC10357475.
We currently exclude the following resources (we plan to later incorporate them):
Variant Curation Expert Panel (VCEP) Criteria Specifications approved by the SVI VCEP Review Committee
Strande NT, Riggs ER, Buchanan AH, Ceyhan-Birsoy O, DiStefano M, Dwight SS, Goldstein J, Ghosh R, Seifert BA, Sneddon TP, Wright MW, Milko LV, Cherry JM, Giovanni MA, Murray MF, O’Daniel JM, Ramos EM, Santani AB, Scott AF, Plon SE, Rehm HL, Martin CL, Berg JS. Evaluating the Clinical Validity of Gene-Disease Associations: An Evidence-Based Framework Developed by the Clinical Genome Resource. Am J Hum Genet. 2017 Jun 1;100(6):895-906. doi: 10.1016/j.ajhg.2017.04.015. Epub 2017 May 25. PMID: 28552198; PMCID: PMC5473734.
Further Supporting Literature
Eilbeck K, Lewis SE, Mungall CJ, Yandell M, Stein L, Durbin R, Ashburner M. The Sequence Ontology: a tool for the unification of genome annotations. Genome Biol. 2005;6(5):R44. doi: 10.1186/gb-2005-6-5-r44. Epub 2005 Apr 29. PMID: 15892872; PMCID: PMC1175956.
Quinodoz M, Royer-Bertrand B, Cisarova K, Di Gioia SA, Superti-Furga A, Rivolta C. DOMINO: using machine learning to predict genes associated with dominant disorders. The American Journal of Human Genetics. 2017 Oct 5;101(4):623-9.
Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Aguilera MA, Meyer R, Massouras A. VarSome: the human genomic variant search engine. Bioinformatics. 2019 Jun 6;35(11):1978.
Martin AR, Williams E, Foulger RE, Leigh S, Daugherty LC, Niblock O, Leong IU, Smith KR, Gerasimenko O, Haraldsdottir E, Thomas E. PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nature genetics. 2019 Nov;51(11):1560-5.
Thormann A, Halachev M, McLaren W, Moore DJ, Svinti V, Campbell A, Kerr SM, Tischkowitz M, Hunt SE, Dunlop MG, Hurles ME. Flexible and scalable diagnostic filtering of genomic variants using G2P with Ensembl VEP. Nature communications. 2019 May 30;10(1):2373.
Gudmundsson S, Singer-Berk M, Watts NA, Phu W, Goodrich JK, Solomonson M; Genome Aggregation Database Consortium; Rehm HL, MacArthur DG, O’Donnell-Luria A. Variant interpretation using population databases: Lessons from gnomAD. Hum Mutat. 2022 Aug;43(8):1012-1030. doi: 10.1002/humu.24309. Epub 2021 Dec 16. PMID: 34859531; PMCID: PMC9160216.
Criteria#
The text in the following section is based on the one by Richards et al. (2015) and the updates listed in References.
Pathogenic Very Strong#
PVS1 (null variant)#
variant is a null variant (sequence ontology:
stop_gained,frameshift_variant,splice_acceptor_variant,splice_donor_variant,start_lost,exon_loss_variant,transcript_variant)loss of function is a known disease mechanism for the affected
incorporate figures 4-5 from Walker et al. (2023)
Caveats
beware of genes where LOF is not a known disease mechanism (e.g., GFAP, MYH7)
caution when interpreting LOF at the extreme 3’ and of gene
caution with splice variants predicted to lead to exon skipping but leave the remainder of the protein intact
caution in the presence of multiple transcripts
PVS1 Update 2018#
Decision Tree
In Tayoun et al. (2018), the following decision tree is defined. It is based on the assumption that the gene-disease association is at a moderate, strong, or definitive clinical level according to Strande et al. (2017). Note that we do not incorporate the matching by Strande et al. (2017) for now.
stop_gainedorframeshift_variant- predicted to undergo NMD
- exon is present in biologically-relevant transcripts
result:
PVS1
- exon is absent from biologically-relevant transcripts
result: N/A
- not predicted to undergo NMD
- truncated / altered region is critical to protein function
result:
PVS_Strong
- role of region in protein function is unknown
- LoF variants in this exon are frequent in the general population and/or exon is absent from biologically-relevant transcripts
result: N/A
- LoF variants in this exon are not frequent in the general population and exon is present in biologically-relevant transcripts
- variant removes >=10% of the protein
result:
PVS_Strong
- variant removes <10% of the protein
result:
PVS1_Moderate
splice_acceptor_variantorsplice_donor_variant- exon skipping or use of a cryptic splice site disrupts reading frame and is predicted to undergo NMD
- exon is present in biologically-relevant transcripts
result:
PVS1
- exon is absent from biologically-relevant transcripts
result: N/A
- exon skipping or use of a cryptic splice site disrupts reading frame and is NOT predicted to undergo NMD
- truncated / altered region is critical to protein function
result:
PVS_Strong
- role of region in protein function is unknown
- LoF variants in this exon are frequent in the general population and/or exon is absent from biologically-relevant transcripts
result: N/A
- LoF variants in this exon are not frequent in the general population and exon is present in biologically-relevant transcripts
- variant removes >=10% of the protein
result:
PVS_Strong
- variant removes <10% of the protein
result:
PVS1_Moderate
- exon skipping or use of a cryptic splice site rpeserves reading frame
- role of region in protein is unknown
- LoF variants in this exon are frequent in the general population and/or exon is absent from biologically-relevant transcripts
result: N/A
- LoF variants in this exon are not frequent in the general population and exon is present in biologically-relevant transcripts
- variant removes >=10% of the protein
result:
PVS_Strong
- variant removes <10% of the protein
result:
PVS1_Moderate
- truncated / altered region is critical to protein function
result:
PVS_Strong
exon_loss_variantortranscript_variant(single exon or whole transcript deletion)- full gene deletion
result:
PVS1
- single to multi exon deletion - disrupts reading frame and is predicted to undergo NMD
- exon is present in biologically-relevant transcripts
result:
PVS1
- exon is absent from biologically-relevant transcripts
result: N/A
- single to multi exon deletion - disrupts reading frame and is NOT predicted to undergo NMD
- truncated/altered region is critical to protein function
result:
PVS_Strong
- role of region in protein function is unknown
- LoF variants in this exon are frequent in the general population and/or exon is absent from biologically-relevant transcripts
result: N/A
- LoF variants in this exon are not frequent in the general population and exon is present in biologically-relevant transcripts
- variant removes >=10% of the eprotein
result:
PVS_Strong
- variant removes <10% of the protein
result:
PVS1_Moderate
- single to multi exon deletion - preserves reading frame
role of region in protein function is unknown – see 3.3.2
- truncated / altered region is critical to protein function
result:
PVS_Strong
- duplication (>=1 exon in size and must be contained within gene)
- proven in tandem
- reading frame disrupted and NMD predicted to occur
result:
PVS1
- no or unknown impact on reading frame and NMD
result: N/!
- presumed in tandem
- reading frame presumed disrupted and NMD predicted to occur
result:
PVS1
- no or unknown impact on reading frame and NMD
result: N/A
- proven not in tandem
result: N/A
start_lost- no known alternative start codon in other transcripts
- >=1 pathogenic variant(s) upstream of closest potential in-frame start codon
result:
PVS1_Moderate
- no pathogenic variant(s) upstream of closest potential in-frame start codon
result:
PVS1_Supp
- different functional transcript uses alternative start codon
result: N/A
And here is the tree as an image:

Notes
criterion (2)
splice_acceptor_variantorsplice_donor_variantis mutually exclusive to splice site prediction“Generally, NMD is not predicted to occur if the premature termination codon occurs in the 3’ most exon or within the 3’ most 50 nucleotides of the penultimate exon” – from Tayoun et al. (2018)
Criteria for LoF Disease Mechanism
Further, Tayoun et al. (2018) define the following criteria for a loss-of-function disease mechanism.
- Follow PVS1 decision tree if:
clinical validaity classification of gene is strong or definite AND
>=3 LoF functions are Pathogenic without PVS1 AND
>10% of variants associated with the phenotype are LoF (must be across more than 1 exon - except for single-exon genes)
- Decrease final strenght by one level (IOW: to
PVS1_Strong) if: clinical validity classification of gene is at least moderate AND
>=2 LoF variants have previously associated with the phenotype (must be across more than one exon - except for single-exon genes) AND
null mouse model recapitulates disease phenotype
- Decrease final strenght by one level (IOW: to
- Decrease final strength by two levels (IOW: to
PVS1_Moderate) if: - clinical validity classification of gene is at least moderate AND EITHER
>=2 LoF variants have been previously associated with the phenotype (must be across more than one exon - except for single-exon genes) OR
null mouse model recapitulates disease phenotype
- Decrease final strength by two levels (IOW: to
If there is no evidence that LoF variants cause disease, PVS1 should not be applied at any strength level.
Pathogenic Strong#
PS1 (same amino acid change)#
same amino acid change has previoulsy been established as pathogenic, regardless of nucleotide change
for splicing variants, Tables 2-3 from Walker et al. (2023) shall be used
Table 2 Rules from Walker et al. (2023) of Variant under assessment (VUA)
- VUA located outside splice donor / acceptor +/- 1/2 dinucleotide positions
- baseline computational code: PP3
- position of comparison variant relative to VUA: same nucleotide
with P comparison variant: PS1
with LP comparison variant: PS1_Moderate
- position of comparison variant relative to VUA: within same splice donor / acceptor motif (including +/- 1/2 position)
with P comparison variant: PS1_Moderate
with LP comparison variant: PS1_Supporting
- VUA located at splice donor / acceptor +/- 1/2 dinucleotide positions
- baseline computational code: PVS1
position of comparison variant relative to VUA: within same splice donor / acceptor +/- 1/2 dinicleotide
with P comparison variant: VUA is PS1_Supporting
with LP comparison variant: N/A
- baseline computational code: PVS1
position of comparison variant relative to VUA: within same splice donor / acceptor region but outside +/- 1/2 dinicleotide
with P comparison variant: VUA is PS1_Supporting
with LP comparison variant: VUA is PS1_Supporting
- baseline computational code: PVS1_Strong, PVS1_Moderate, PVS1_Supporting
position of comparison variant relative to VUA: within same splice donor / acceptor +/- 1/2 dinicleotide
with P comparison variant: VUA is PS1
with LP comparison variant: VUA is N/A
- baseline computational code: PVS1_Strong, PVS1_Moderate, PVS1_Supporting
position of comparison variant relative to VUA: within same splice donor / acceptor motify but outside +/- 1/2 dinicleotide
with P comparison variant: VUA is PS1_Moderate
with LP comparison variant: VUA is PS1_Supporting
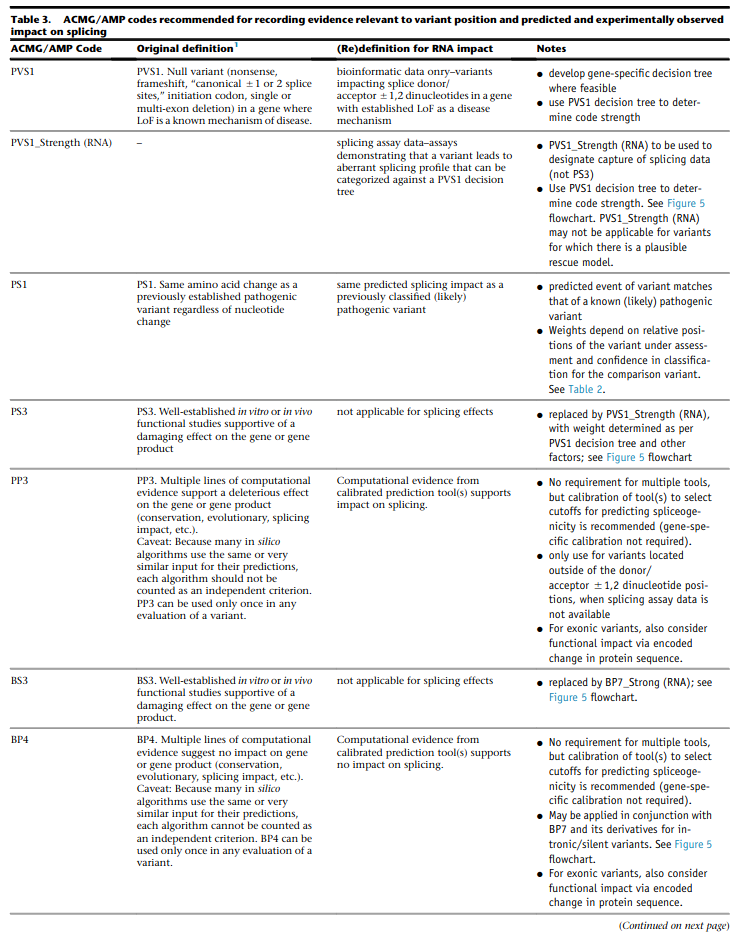
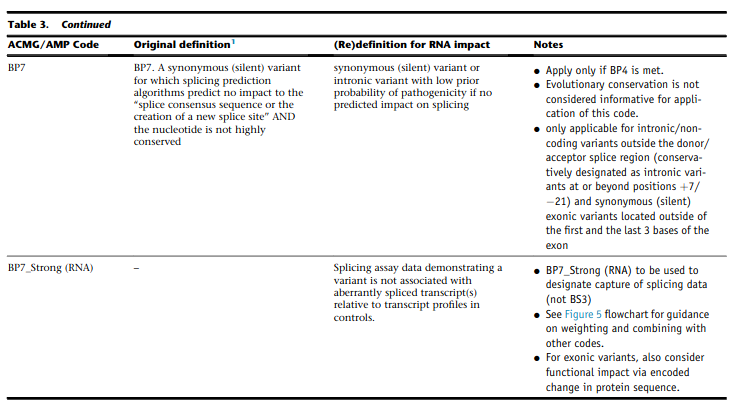
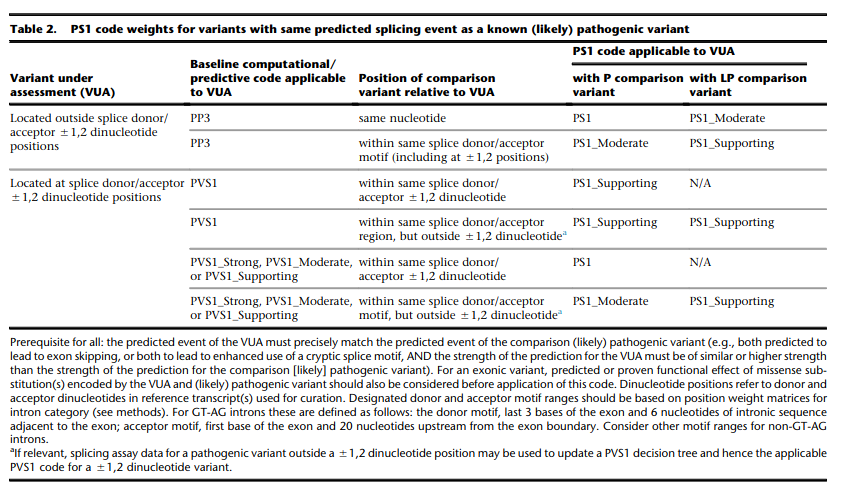
Caveats
beware of changes that impact splicing rather than amino acid or protein level
Notes
incorporation of splicing here is based on the recommendation by Walker et al. (2023)
PS2 (confirmed de novo)#
confirmed de novo variant in a patient withou disease and no family history
Caveats
confirmation of paternity only is insufficient (egg donation, surrogate motherhood, errors in embryo transfer, … can contribute to nonmaternity)
Notes
ClinGen Sequence Variant Interpretation Work Group (2021) describe a point-scale for PS2 and PM6. However, this is hard to apply automatically as it requires an assessment of whether the phenotype is highly specific or consistent with the gene.
PS3 (functional studies)#
well-established in vitro or in vivo functional studies supportive of a damaging effect on the gene or gene product
Caveats
functional studies that have been validated and shown to be reproducible and robust in a clinical diagnostic laboratory setting are considered the most well established
Notes
There is further guidance in Brnich et al. (2020) on how to apply PS3 and BS3 when interpreting “well-established” functional assays.
However, as this process is manual, it is not further considered here.
Walker et al. (2023) is not considered here as the authors recommend to capture experimental evidence with PVS1 and is not suitable for automatic classification.
PS4 (prevalence)#
prevalence of the variant in affected individuals is significantly increased compared with the prevalence in controls
Caveats
relative risk or OR, as obtained from case–control studies, is >5.0, and the confidence interval around the estimate of relative risk or OR does not include 1.0. See Richards et al. (2015) for detailed guidance.
in instances of very rare variants where case–control studies may not reach statistical significance, the prior observation of the variant in multiple unrelated patients with the same phenotype, and its absence in controls, may be used as moderate level of evidence.
Pathogenic Moderate#
PM1 (hotspot)#
located in a mutational hot spot and/or critical and well-established functional domain (e.g., active site of an enzyme) without benign variation
Caveats
Pejaver et al. (2022) suggest to limit combined evidence from P1 and PP3 to strong
PM3 (recessive and in trans)#
for recessive disorders, detected in trans with a pathogenic or likely pathogenic variant in an affected patient
According to ClinGen Sequence Variant Interpretation Work Group (2019), there are points awarded per in trans proband (all variants should be sufficiently rare, thus meet PM specifiacation, P-Pathogenic or LP-Likely pathogenic):
Classification / zygosity of other variant |
Points per confirmed in trans |
Points if phase unknown |
Pathogenic or Likely pathogenic variant |
1.0 |
0.5(P) or 0.25(LP) |
Homozygous occurence (max point 1.0) |
0.5 |
N/A |
Uncertain significance variant |
0.25 |
0.0 |
The resulting point rating gives the following evidence strength for PM3:
0.5-1.0: PM3_Supporting
1.0-2.0: PM3
2.0-4.0: PM3_Strong
>=4.0: PM3_VeryStrong
Notes
ClinGen Sequence Variant Interpretation Work Group (2019) changes this from “for recessive disorders, detected in trans with a pathogenic” to “for recessive disorders, detected in trans with a pathogenic or likely pathogenic variant in an affected patient”
Further, this document introduces the point-based system from above.
There are further considerations in ClinGen Sequence Variant Interpretation Work Group (2019) that are not considered here.
PM4 (protein length)#
protein length changes as a result of in-frame deletions/insertions in a nonrepeat region or stop-loss variants
PM5 (overlapping missense)#
novel missense change at an amino acid residue where a different missense change determined to be pathogenic has been seen before
Caveats
beware of changes that impact splicing rather than at the amino acid/protein level.
PM6 (assumed de novo)#
assumed de novo, but without confirmation of paternity and maternity
Pathogenic Supporting#
PM2_Supporting (absent from controls)#
absent from controls (or at extremely low frequency if recessive) in gnomAD
Notes
population indel data is of high quality by now
ClinGen Sequence Variant Interpretation Work Group (2020) has downgraded this to PM2_Supporting by default.
PP1 (cosegregaton)#
cosegregation with disease in multiple affected family members in a gene definitively known to cause th disease
Notes
may be used as stronger evidence with increasing segregation data
PP2 (missense)#
missense variant in a gene that has a low rate of benign missense variation and in which missense variants are a common mechanism of disease
PP3 (in silico predictions)#
multiple lines of computational evidence support a deleterious effect on the gene or gene product (conservation, evolutionary, splicing impact, etc.)
incorporate figures 4-5 from Walker et al. (2023)

Caveats
because many in silico algorithms use the same or very similar input for their predictions, each algorithm should not be counted as an independent criterion. PP3 can be used only once in any evaluation of a variant.
Pejaver et al. (2022) suggest to limit combined evidence from P1 and PP3 to strong.
Notes
The resulting class is updated according to the Pejaver et al. (2022). Note that it would be very useful to run the original code by Pejaver with more scores. The code from Pejaver can be found here on GitHub.
PP4 (monogenetic)#
patient’s phenotype or family history is highly specific for a disease with a single genetic etiology
PP5 (reputable source)#
Remove according to Biesecker et al. (2018).
Benign Standalone#
BA1 (5% frequency)#
allele frequency is >5% in Exome Sequencing Project, 1000 Genomes Project, or Exome Aggregation Consortium
In accordance with Ghosh et al. (2018), there is a list of exceptions from this rule with high MAF but some evidence for pathogenicity. Updates to this list are available at ClinGen and shall be monitored regularly.
NM_014049.4(ACAD):c.-44_-41dupTAAGNM_004004.5(GJB2):c.109G>A (p.Val37Ile)NM_000410.3(HFE):c.187C>G (p.His63Asp)NM_000410.3(HFE):c.845G>A (p.Cys282Tyr)NM_000243.2(MEFV):c.1105C>T (p.Pro369Ser)NM_000243.2(MEFV):c.1223G>A (p.Arg408Gln)NM_006346.2(PIBF1):c.1214G>A (p.Arg405Gln)NM_000017.3(ACADS):c.511C>T (p.Arg171Trp)NM_000060.4(BTD):c.1330G>C (p.Asp444His)
Benign Very Strong#
This category does not exist in Richards et al. (2015) but is implicitely introduced by Tavtigian et al. (2020).
Benign Strong#
BS1 (expected frequency)#
allele frequency is greater than expected for disorder
BS2 (healthy adult)#
observed in a healthy adult individual for a recessive (homozygous), dominant (heterozygous), or X-linked (hemizygous) disorder, with full penetrance expected at an early age
BS3 (functional studies)#
well-established in vitro or in vivo functional studies show no damaging effect on protein function or splicing
Notes
There is further guidance in Brnich et al. (2020) on how to apply PS3 and BS3 when interpreting “well-established” functional assays. However, as this process is manual, it is not further considered here.
Walker et al. (2023) is not considered here as the authors recommend to capture experimental evidence with PVS1 and is not suitable for automatic classification.
BS4 (lack of segregation)#
lack of segregation in affected members of a family
Caveat
presence of phenocopies for common phenotypes (i.e., cancer, epilepsy) can mimic lack of segregation among affected individuals
families may have more than one pathogenic variant contributing to an autosomal dominant disorder, further confounding an apparent lack of segregation
Benign Moderate#
This category does not exist in Richards et al. (2015) but is implicitely introduced by Tavtigian et al. (2020).
Benign Supporting#
BP1 (missense)#
missense variant in a gene for which primarily truncating variants are known to cause disease
BP2 (in trans)#
Observed in trans with a pathogenic variant for a fully penetrant dominant gene/disorder or observed in cis with a pathogenic variant in any inheritance pattern
BP3 (in-frame in repetitive)#
in-frame deletions/insertions in a repetitive region without a known function
BP4 (in silico predictions)#
multiple lines of computational evidence suggest no impact on gene or gene product (conservation, evolutionary, splicing impact, etc.)
incorporate figures 4-5 from Walker et al. (2023)
Caveats
because many in silico algorithms use the same or very similar input for their predictions, each algorithm cannot be counted as an independent criterion. BP4 can be used only once in any evaluation of a variant.
Notes
The resulting class is updated according to the Pejaver et al. (2022). Note that it would be very useful to run the original code by Pejaver with more scores. The code from Pejaver can be found here on GitHub.
BP5 (found in solved)#
variant found in a case with an alternate molecular basis for disease
BP6 (reputable source)#
Remove according to Biesecker et al. (2018).
BP7 (synonymous)#
synonymous (silent) variant for which splicing prediction algorithms predict no impact to the splice consensus sequence nor the creation of a new splice site AND the nucleotide is not highly conserved
incorporate figures 4-5 from Walker et al. (2023)